Edit 10/9/14 – Updated statistics for the 1024 bit fingerprints
There’s been some discussion about a paper by O’Hagan et al that have proposed a Rule of 0.5 that states that 90% of approved drugs exhibit a Tanimoto similarity > 0.5 to one or more human metabolites. Their analysis is based on metabolites listed in Recon2, a reconstruction of the human metabolic network. The idea makes sense and there’s an in depth discussion at In the Pipeline.
Given the authors’ claim that
a successful drug is likely to lie within a Tanimoto distance of 0.5 of a known human metabolite. While this does not mean, of course, that a molecule obeying the rule is likely to become a marketed drug for humans, it does mean that a molecule that fails to obey the rule is statistically most unlikely to do so
I was interested in seeing how this rule of thumb holds up when faced with compounds that are not supposed to make it through the drug development pipeline. Since PAINS appear to be the structural filter du jour, I decided to look at compounds that failed the PAINS filter. I worked with the 10,000 compounds included in Saubern et al. Simon Saubern provided me the set of 861 compounds that failed the PAINS filters, allowing me to extract the set of compounds that passed (9139)
Chris Swain was kind enough to extract the compound entries from the Matlab dump provided by O’Hagan et al. This file contained InChI representations for a subset of the entries. I extracted the 2980 valid InChI strings and converted them to SMILES using ChemAxon molconvert 6.0.5. The processed data (metabolite name, InChI and SMILES) are available here. However, after deduplication, there were 1335 unique metabolites
Now, O’Hagan et al for some reason, used the 166 bit MACCS keys, but hashed them to 1024 bits. Usually, when using a keyed fingerprint, the goal is to retain the correspondence between bit position and substructure. The hashing step results in a loss of such correspondence. So it’s a bit surprising that they didn’t use some sort of path (Daylight) or environment (ECFPn) based fingerprint. Since I didn’t know how they hashed the MACCS keys, I calculated 166 bit MACCS keys and 1024 bt ECFP6 and extended path fingerprints using the CDK (via rcdk). Then for each compound in the PAINS pass or fail set, I computed the similarity to each of the 1335 metabolites and identified the maximum similarity (termed NMTS in the paper) and then plotted the distribution of these NMTS values between the PAINS pass and fail sets.
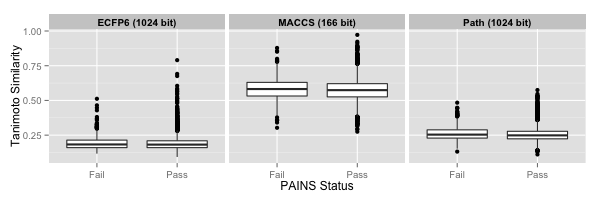
First, the similarity cutoff proposed by the authors is obiously dependent on the fingerprint. So while the bulk of the 166 bit MACCS similarities are > 0.5, this is not really meaningful. A more relevant comparison is to 1024 bit fingerprints – both are hashed, so should be somewhat comparable to the authors choice of hashed MACCS keys.
The path fingerprints lead to an NMTS of ~ 0.25 for both PAINS pass and fail sets and the ECFP6 leads to an NMTS of ~ 0.18 for both sets. Though the difference in medians between the pass and fail sets for the path fingerprint is statistically significant (p = 1.498e-05, Wilcoxon test), the difference itself is very small: 0.005. (For the circular fingerprint there is no statistically significant difference). However, the PAINS pass set does contain more outliers with values > 0.5. In that sense the proposed rule does separate the two groups. Of the top of my head I don’t know whether the WEHI screening deck that was the source of the 10,000 compounds was designed to be drug-like. At the same time all this might be saying is there is no relationship between metabolite-likenes and PAINS-likeness.
It’d be interesting to see how this type of analysis holds up with other well known filter rules (REOS, Lilly etc). A related thing to look at would be to see how druglikeness scores compare with NMTS values.
Code and data are available in this repository
Raj
Not that I’m a fan of either one, but…
… it’s not surprising that there is no correlation between the two models. One is supposed to be predict potential tox issues, the other predicts assay interference.